
1. The Intellectual Prelude: Foundations of Molecular Replication
The development of the Polymerase Chain Reaction (PCR) represents a masterclass in how foundational scientific discovery serves as a prerequisite for disruptive commercial innovation. In the mid-20th century, the molecular biology landscape was defined by the elucidation of DNA’s structure and the biochemical rules governing its replication. For a strategist, these were not merely academic triumphs; they established the “technical floor” upon which PCR would be built. The understanding of complementary base pairing and the isolation of DNA-synthesizing enzymes provided the necessary blueprints for high-throughput DNA manipulation. Without this structural and enzymatic context, the eventual “chain reaction” concept would have lacked both the theoretical logic and the biochemical tools required to achieve exponential amplification.
The following milestones established the strategic and conceptual scaffolding for PCR:
• 1953: The Double Helix (Watson and Crick): Founders of molecular genetics, their model of complementary base-paired DNA strands immediately suggested a biological copying mechanism. This established the fundamental logic of using one strand as a template for another.
• 1957: Discovery of DNA Polymerase I (Arthur Kornberg): Kornberg isolated the first “engine” of replication. While he received the Nobel Prize in 1959, the early enzyme was limited by its one-directional synthesis and requirement for an existing primer.
• 1960s: Synthetic Oligonucleotides (H. Gobind Khorana): Khorana pioneered the synthesis of DNA fragments. These oligonucleotides were the strategic precursors to the primers that would eventually allow researchers to “target” specific genomic regions.
• 1971: The Two-Primer Vision (Kjell Kleppe): Working in Khorana’s laboratory, Kleppe envisioned a system using two primers to replicate a specific DNA segment. While his “repair replication” approach did not achieve exponential growth, it provided the first clear theoretical description of the PCR principle.
Pre-PCR Technical Components
| Component | Historical Origin / Discoverer |
|---|---|
| DNA Polymerase I | Arthur Kornberg (1957) |
| Klenow Fragment | Klenow (1970) |
| DNA Polymerase from T. aquaticus | Chien et al. (1976) |
| Synthetic Oligonucleotides | H. Gobind Khorana (1960s) |
By the dawn of the 1980s, the theoretical pieces were commercially ripe, shifting the locus of innovation from academic laboratories to the emerging corporate environment of Cetus Corporation in Emeryville, California.
2. The “Molecular Copier”: An Introduction to DNA Amplification
Imagine you are a detective trying to find a single, specific sentence printed on a page the size of a football field. Before 1983, this was the central frustration of molecular biology. Scientists often possessed the “book of life,” but they lacked the magnifying glass to read it. They frequently found themselves with too little DNA to study; a tiny biological trace was essentially invisible to the equipment of the time. The challenge was not just seeing the DNA, but having enough of it to experiment with, sequence, or manipulate.
The “Eureka!” moment that solved this occurred not in a sterile lab, but on the winding Pacific Coast Highway. In May 1983, a chemist named Kary Mullis was driving through the California mountains when he conceived of the Polymerase Chain Reaction (PCR). He realized that by using two “primers” to bookend a specific sequence, he could trigger a chain reaction that would copy that sequence over and over again. PCR is, quite literally, a “molecular copier.” It allows a scientist to take a single segment of DNA and, in just a few hours, produce millions or billions of identical copies, bridging the gap between a microscopic trace and a major discovery.
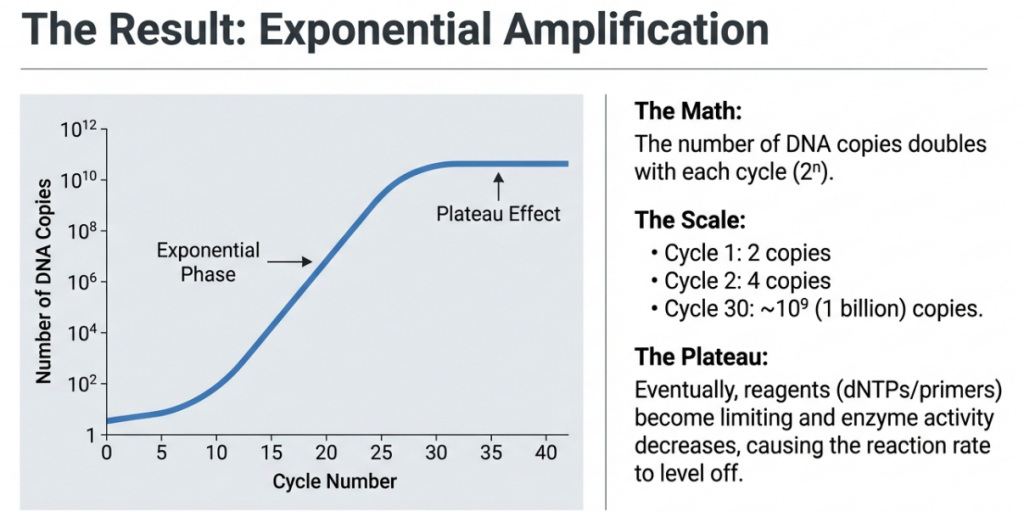
Key Concept: Exponential Growth PCR operates on a principle of geometric amplification. Because the products of one cycle become the templates for the next, the DNA doubles every round. The mathematical formula for this is 2n, where n is the number of cycles.
Visualizing the Growth:
• Cycle 0: == (1 copy)
• Cycle 1: == == (2 copies)
• Cycle 2: == == == == (4 copies)
• Cycle 3: == == == == == == == == (8 copies)
• Cycle 4: (16 copies)… and so on.
By Cycle 30, you have over 1 billion copies of your target! However, the reaction eventually hits a plateau. Just as a real copy machine eventually “runs out of ink,” the PCR tube eventually runs out of free dNTP “bricks” and primers, causing the growth to level off. While the math is elegant, the physical execution was originally a “tedious and costly” nightmare. To understand why it became the foundation of modern biology, we must look at the rhythmic “molecular dance” of the reaction itself.
3. The Three-Step Dance: How PCR Works
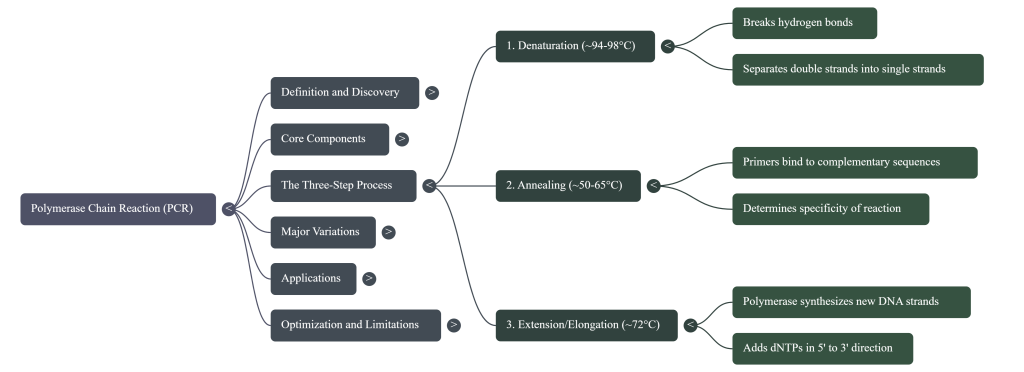
In nature, DNA strands are “zipped up” tight for protection. To copy them, we must use temperature to unlock the double helix and guide our molecular builder. PCR moves the reaction through three distinct stages, forming one “thermal cycle.”
1. Denaturation (The Heat): The reaction is heated to approximately 94–98°C. At this near-boiling temperature, the hydrogen bonds holding the two strands together break. The DNA double helix “unzips,” resulting in two single strands of DNA ready to be copied.
2. Annealing (The Cool Down): The temperature is lowered to roughly 50–65°C. This allows the short primers to find their matching matches on the single strands and “stick” to them. This step must be precise: if it’s too hot, the primers won’t stick; if it’s too cool, they might stick to the wrong places.
3. Extension (The Building Phase): The temperature is shifted to approximately 72°C. While 75–80°C is technically the biological optimum for the Taq enzyme, 72°C is the lab standard for stability. Here, the polymerase binds to the primers and starts adding dNTPs to synthesize a brand-new complementary arm of DNA.
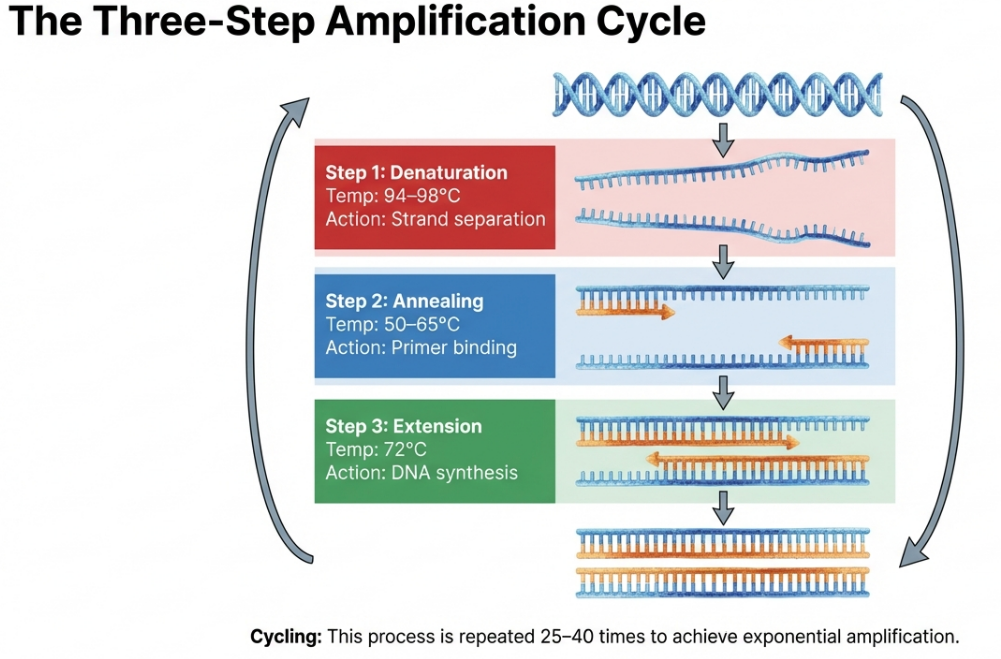
This single cycle is just the beginning of a massive chain reaction that grows at a staggering rate.
| Step Name | Temperature Range | What Happens to the DNA |
|---|---|---|
| Denaturation | 94°C – 98°C | The high heat acts like a “molecular zipper,” breaking the hydrogen bonds holding the two strands together to create single-stranded templates. |
| Annealing | 50°C – 65°C | The temperature drops to allow primers to find their targets through complementary base pairing, precisely “bookending” the sequence to be copied. |
| Extension | 70°C – 80°C | The polymerase enzyme builds new DNA by adding building blocks (dNTPs) to the primers, synthesizing a fresh complementary strand. |
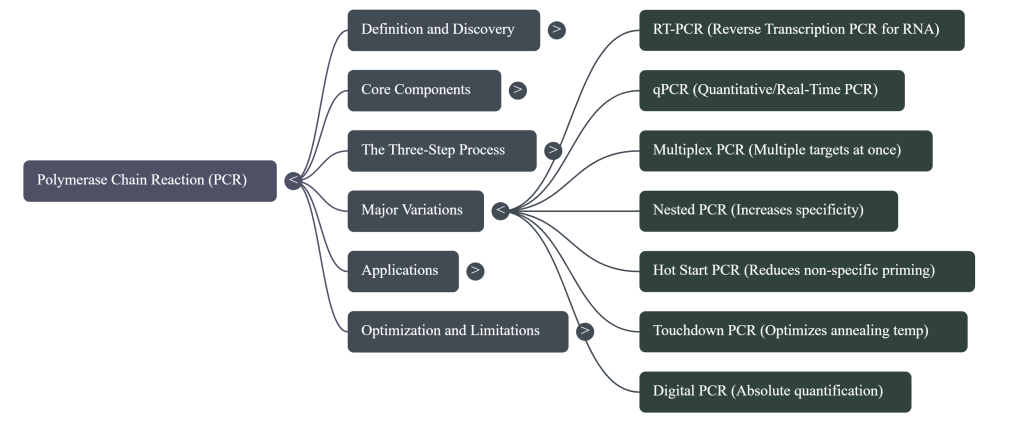
The “Magic Ingredient”: Taq Polymerase
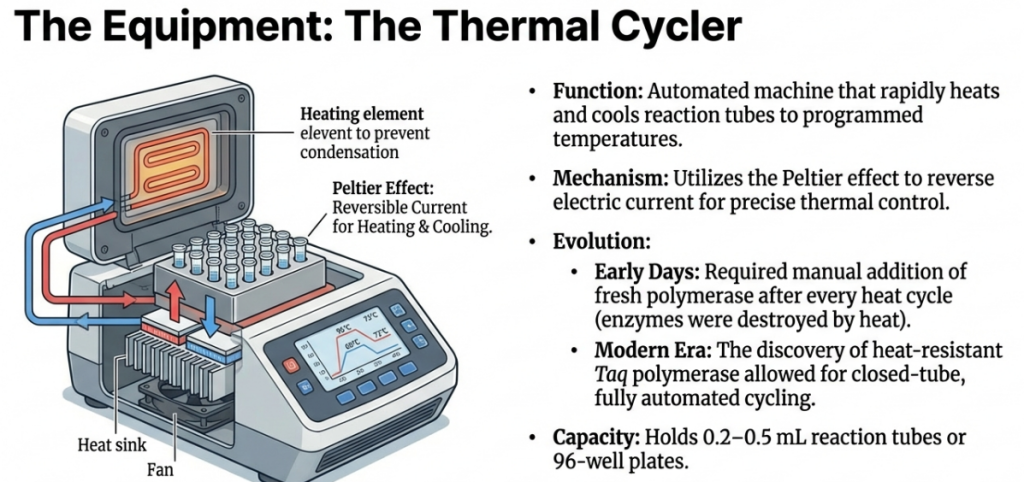
In the early days, PCR was a manual labor of love. Scientists used the Klenow fragment (from E. coli), which was destroyed by the high heat of the denaturation step. This meant a researcher had to stand over the machine and manually add fresh enzyme every single cycle. The revolution came with Taq Polymerase, isolated from Thermus aquaticus, a bacterium found in the superheated Mushroom Spring of Yellowstone National Park. Because this enzyme is heat-stable, it survives the “melting” phase, allowing the entire process to be automated in a machine called a thermal cycler.
The choice of DNA polymerase determines the analytical speed, yield, and accuracy of the assay:
• Taq Polymerase: Isolated from Thermus aquaticus, this is the industry standard for processivity and speed (typically 1 kb/min). It is highly thermostable and exhibits terminal transferase activity, adding a single 3′ adenine (A) overhang to products.
• Pfu Polymerase: Derived from Pyrococcus furiosus, this enzyme is selected when high-fidelity sequence accuracy is non-negotiable. Its 3′ to 5′ exonuclease proofreading activity significantly reduces error rates, though it requires longer extension times (approximately 2 min/kb).
Expert Tip The discovery of Taq was the missing link that allowed for the automation of PCR. It allowed engineers to build the thermal cycler, a machine that automatically cycles through temperatures so scientists can “set it and forget it.”
The PCR “Master Mix”
Think of a PCR reaction as a precision construction project. You wouldn’t show up to a job site without your blueprints, your workers, and your bricks. In the molecular world, we assemble a similar chemical “toolkit” in a small tube. Each ingredient is carefully chosen to ensure that we build only the specific segment of DNA we are looking for.
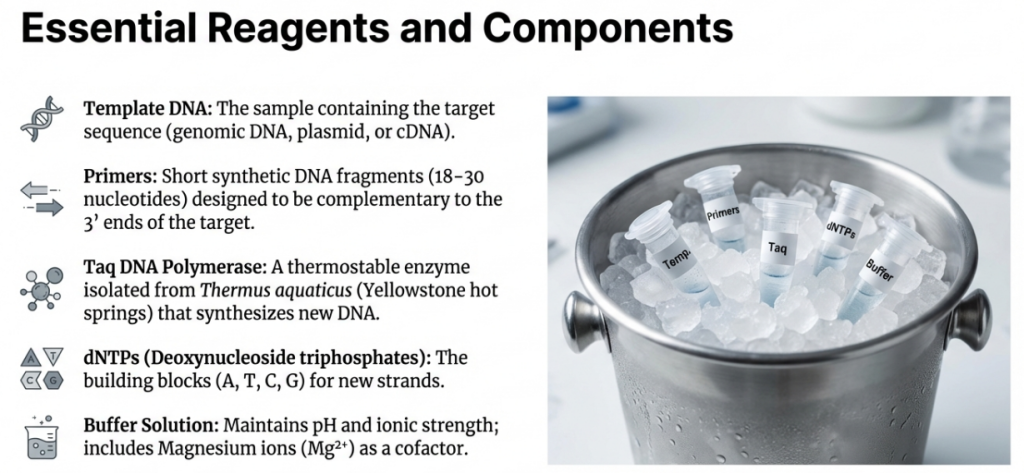
| Ingredient | Role in the Reaction | Everyday Analogy |
|---|---|---|
| DNA Template | The original sample containing the specific target sequence you want to copy. | The Blueprint: The original document you want to duplicate. |
| Primers (Oligonucleotides) | Short, synthetic DNA fragments that mark the beginning and end of the target section. | “Start Here” Signs: They tell the machine exactly which part of the blueprint to copy. |
| Taq Polymerase | A specialized enzyme that acts as a worker, physically assembling the new DNA strands. | The Builder: The construction worker who reads the blueprint and lays the bricks. |
| dNTPs (Deoxynucleotides) | The four individual building blocks (A, T, C, and G) that make up DNA. | The Bricks: The raw materials used to build the new structure. |
| Buffer & Salts (Mg2+ / K+) | A solution containing Magnesium (for the enzyme) and Potassium (to help DNA strands separate). | The Life Support System: A “power drink” environment that keeps the builder at peak performance. |
Ions and Substrates
• Bivalent Cations (Mg²⁺, Mn²⁺): Magnesium is an essential cofactor for thermostable polymerases. A critical insight for optimization is that Mg²⁺ is not consumed during the reaction; however, its availability is heavily buffered by the concentration of dNTPs. Mn²⁺ may be substituted in specific mutagenic applications to intentionally increase error rates.
• Monovalent Cations (K⁺): Potassium ions (35–100 mM) stabilize primer annealing. Titrating KCl allows for the preferential denaturation of shorter DNA molecules over longer ones, a useful tactic for improving specificity in amplicons under 1000 bp.
• dNTPs: Deoxynucleotide 5′-triphosphates must be maintained in equivalent concentrations ([A]=[T]=[C]=[G]). Because dNTPs chelate Mg²⁺, any significant adjustment in dNTP concentration necessitates a proportional recalibration of Mg²⁺ levels.
Template Integrity
Template purity is a primary determinant of success. Diagnostic assays require high-purity DNA (ideal OD260/280 ratio between 1.8 and 2.0). Common contaminants—including proteins, organic solvents, and detergents like SDS—act as potent polymerase inhibitors that must be identified and removed during the extraction phase.
Primer Architecture
Primer design remains the primary determinant of assay specificity and the most frequent point of failure in molecular diagnostics.
Design Criteria Checklist
Senior scientists must adhere to the following non-negotiable structural constraints:
• Length: 15–30 bases to ensure unique targeting within the genome.
• G-C Content: 40–60%.
• Tm Balance: The melting temperature (Tm) of the primer pair must differ by no more than 5°C.
• 3′ End “G-C Clamp”: The inclusion of a G or C at the 3′ end prevents “breathing”—defined as the fraying or splitting apart of ends due to weak hydrogen bonding. This increases overall priming efficiency.
• Sequence Homogeneity: Mandate the avoidance of di-nucleotide repeats and single base runs (>4 bases) to prevent polymerase slipping and secondary structure formation.
Secondary Structure Risks
Failures often arise from competitive artifacts that exhaust reagents:
• Hairpin Loops: Internal folds caused by self-annealing that render the primer unavailable for template binding.
• Primer-Dimers: Result from 3′ complementarity between primers, leading to the synthesis of small (<100 bp) artifacts that dominate the reaction.
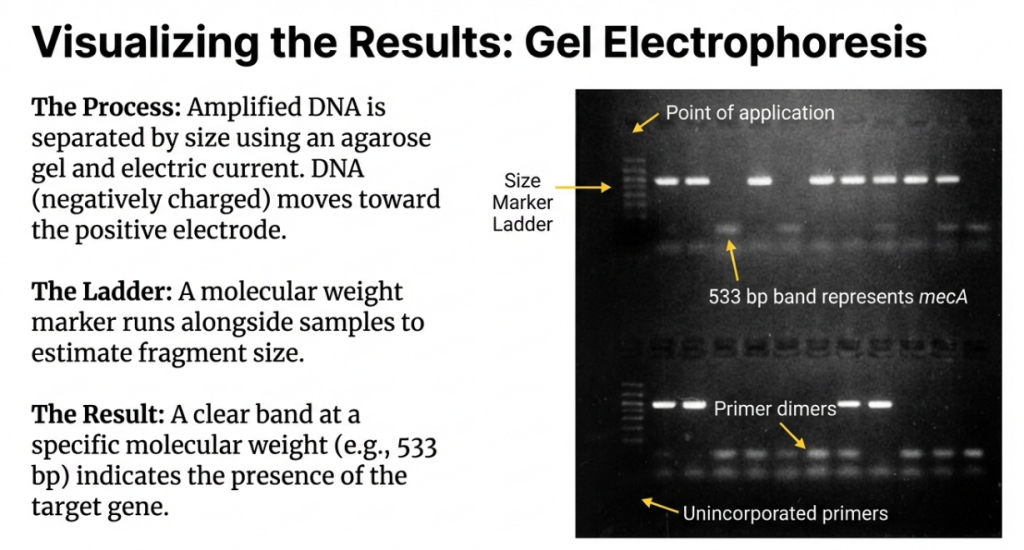
4. Failure Modes: Identification and Root Cause Analysis
Failure Mode Matrix
| Observed Result | Likely Cause | Strategic Response |
|---|---|---|
| No Product Formed | Excessive stringency; missing reagents; inhibitory contaminants (SDS, proteins); insufficient Mg²⁺. | Decrease annealing temperature; check reagent integrity; titrate Mg²⁺; purify template. |
| Non-specific Products (Ladder/Smear) | Low stringency; annealing temperature too low; repetitive sequence targeting; excessive cycles (>35). | Increase annealing temperature; reduce cycle number; titrate KCl. |
| Primer-Dimer (<100bp bands) | High primer-to-template ratio; 3′ complementarity; room-temperature setup. | Reduce primer concentration; use Hot Start PCR; redesign primers. |
| Unintentional Mutations | Use of low-fidelity enzyme (Taq); excessive cycle numbers; high Mg²⁺ or Mn²⁺. | Switch to high-fidelity enzyme (Pfu); reduce Mg²⁺; decrease total cycles. |
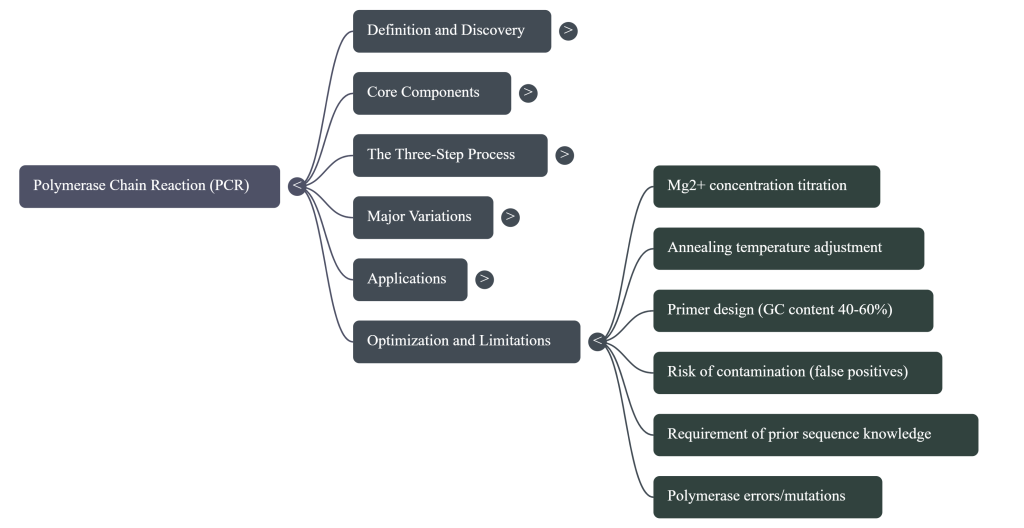
Strategic Optimization of Reagents and Additives
Optimization requires a rigorous titration methodology where only one variable is manipulated at a time to maintain analytical control.
The Mg²⁺ Titration Protocol
Magnesium chloride (MgCl₂) titration (0.5 to 5.0 mM) is the most impactful optimization step. While higher Mg²⁺ levels improve yield, they also stabilize the DNA duplex, which may prevent complete denaturation and encourage spurious annealing to non-target sites.
Enhancer Utility and G-C Rich Templates
Templates with >60% G-C content pose significant challenges. Specialized additives are required:
• DMSO (1-10%) and Betaine (0.5-2.5 M): These disrupt base pairing to lower Tm. Betaine is particularly effective at reducing Tm dependence on nucleotide composition.
• dc7GTP: For extremely difficult G-C templates, incorporate 7-deaza-2′-deoxyguanosine 5′-triphosphate at a 3:1 ratio with dGTP (e.g., 37.5 µM dc7GTP to 12.5 µM dGTP) to destabilize inhibitory secondary structures.
• Formamide (1.25-10%): Increases stringency to reduce non-specific priming.
Neutralizing Inhibitors
When working with environmental samples or degraded DNA, inhibitors must be addressed:
• BSA (Bovine Serum Albumin): Use at 400 ng/µl to alleviate inhibitors like fulvic, humic, or tannic acids, and hemin.
• T4 Gene 32 Protein: Employed at 150 ng/µl as a co-inhibitor neutralizer.
• Refractory Inhibitors: Note that BSA and T4 gene 32 protein cannot neutralize bile salts, SDS, Triton X-100, bilirubin, NaCl, or EDTA. These require further template purification.
Pragmatic Rule: If titration of reagents and additives fails to produce a discrete band, the specialist must redesign the primers and “try, try again.”
Mandatory Laboratory Workflow
Strict adherence to contamination control is non-negotiable to prevent amplicon carry-over:
1. Mandate a Unidirectional Workflow: Movement must be strictly from pre-PCR preparation areas to post-PCR analysis environments.
2. Strict Segregation: Physically separate the reagent preparation room from the product analysis room. Dedicate specific pipettors and equipment to each zone.
3. Decontamination: Use disposable plasticware and perform thorough chemical cleaning of work surfaces between every setup.
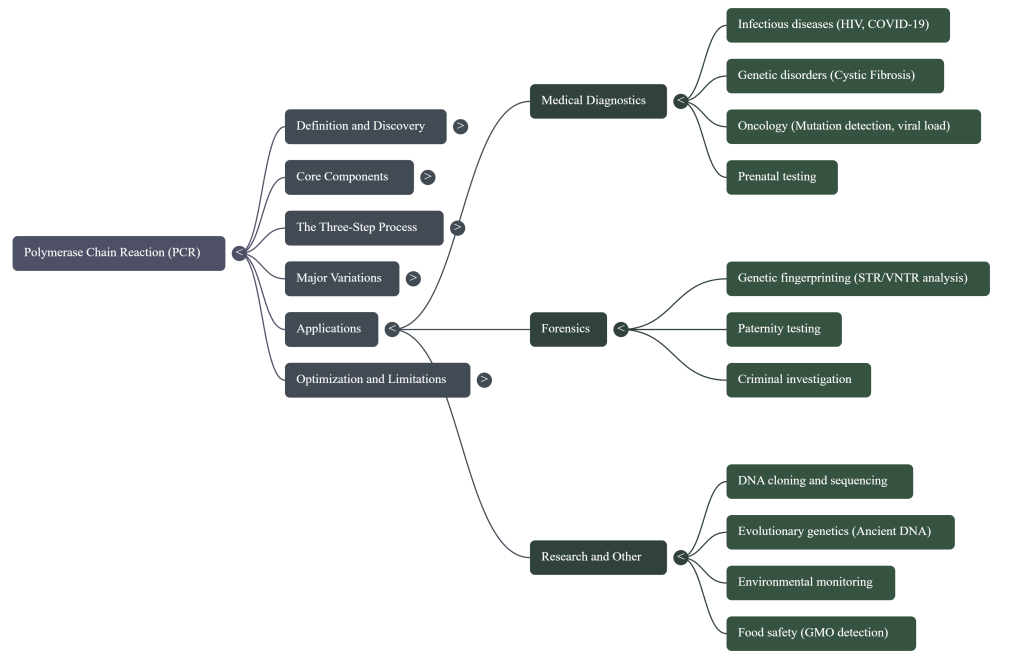
5.1 Application Field Map I: The Medical Frontier
PCR transformed medicine from a reactive field into one of proactive, high-precision diagnostics.
• Infectious Disease Screening
◦ HIV Detection: PCR is so sensitive it can detect as little as one viral genome among 50,000 host cells.
◦ Patient Benefit: This allows for the screening of donated blood long before antibodies appear and enables the immediate testing of newborns.
• Genetic Testing & Personalized Medicine
◦ Sickle Cell Anemia: PCR was first applied clinically to analyze mutations in β-globin genomic sequences for this disorder.
◦ Oncogenes: Doctors use PCR to identify specific mutations in cancer cells.
◦ Patient Benefit: By identifying genetic “triggers,” therapy regimens can be customized to the individual, increasing treatment effectiveness.
5.2 Application Field Map II: The Forensic Revolution
Before PCR, forensic science required large samples of blood or tissue. Today, we can find justice in “trace evidence” that was previously invisible.
• Trace Evidence: A single hair follicle, a few skin cells from under a fingernail, or a minute drop of dried blood provides enough material for analysis.
• The Genetic Fingerprint: PCR targets repetitive, non-coding regions called Short Tandem Repeats (STRs) or Variable Number Tandem Repeats (VNTRs). Every individual (except identical twins) has a unique number of these repeats.
• CODIS (Combined DNA Index System): This is the “national library” of DNA profiles. PCR creates a digital signature of an individual’s STRs that can be compared against this massive database to match crime scene evidence to known suspects.
• Historical Milestones: The 1986 Pennsylvania v. Pestinikas case marked the first criminal use of PCR, and the technique famously played a central role in the evidence analysis of the O.J. Simpson trial.
5.3 Application Field Map III: Archaeology and the Secrets of Ancient DNA
PCR acts as a molecular “time machine,” allowing paleogeneticists to study life that has been dead for millennia.
• Reading Degraded History: DNA breaks down over time, but PCR can seek out the few surviving fragments and amplify them until they are readable.
• Famous Discoveries: PCR was used to identify the body of King Richard III, study Egyptian mummies, and sequence the DNA of a 40,000-year-old mammoth found in ice.
Historical Case Study: “Suicide PCR” When studying ancient pathogens like Yersinia pestis (the plague), scientists extracted DNA from the dental pulp of 14th-century victims. To prevent modern contamination from ruining the results, they used “Suicide PCR.” In this method, a specific primer set is used only once in the lab and never again. This ensures that a positive result is truly from the 14th-century sample and not a “stowaway” from a previous modern experiment.
6. Conclusion: The Foundation for Modern Discovery
Kary Mullis’s drive on the Pacific Coast Highway eventually earned him the Nobel Prize in 1993. However, the true legacy of PCR is that it served as the fundamental technology that allowed the Human Genome Project to be realized. It turned biology into an information science.
Takeaways: Why PCR Matters
1. Precision: It finds the “needle in a haystack” and then builds a whole new haystack out of that one needle.
2. Versatility: It is the primary tool in hospitals, crime labs, and archaeological digs alike.
3. Foundation: Almost every advanced genomic technique, from sequencing to gene editing, begins with a PCR reaction.
Glossary of Key Terms
• Primer: A short synthetic DNA fragment that selects the segment to be amplified.
• Template: The original DNA sample containing the target sequence.
• Thermal Cycler: The laboratory machine that automates the heating and cooling cycles.
• Taq Polymerase: A heat-stable enzyme from Thermus aquaticus that synthesizes new DNA.
• dNTPs: Deoxynucleotide triphosphates; the four building blocks (A, T, C, G) of DNA.
• Magnesium (Mg2+): A metallic ion that acts as an essential cofactor for the DNA polymerase enzyme.
• Amplicon: The final product of a PCR reaction; the millions of identical DNA copies.
Image Summary

Questions/Answers
1. What essential reagents and specialized enzymes are required to execute a PCR reaction?
Executing a Polymerase Chain Reaction (PCR) requires several core ingredients and specialized enzymes that work together to synthesize millions of copies of a specific DNA sequence. These components are typically mixed in a “Master Mix” and placed in a thermal cycler to undergo repeated cycles of temperature changes.
Core Reagents
The fundamental reagents required for a standard PCR include:
• DNA Template: This is the sample containing the target sequence to be amplified. While PCR is sensitive enough to work with the DNA from a single cell, optimal reactions typically use 104 to 107 molecules (roughly 10–500 ng) of purified DNA.
• Primers: These are a pair of short, synthetic single-stranded DNA fragments (oligonucleotides), typically 18–30 nucleotides long. They are designed to be complementary to the 3′ ends of the target DNA region, providing the initiation site for the DNA polymerase.
• Deoxynucleoside Triphosphates (dNTPs): These are the building blocks (A, T, G, and C) used to synthesize new DNA strands. They are typically added in excess to ensure they are not a limiting factor during the reaction.
• Buffer Solution: This reagent maintains the proper pH and ionic strength necessary for the DNA polymerase to remain active and stable. It often includes Tris-HCl and maintains a pH around 9.0.
• Magnesium Ions (Mg2+): Usually added as magnesium chloride (MgCl2), these ions act as a critical cofactor for the DNA polymerase enzyme. Adjusting magnesium concentration is a common method for optimizing reaction yield and specificity.
• Potassium Ions (K+): Typically provided as potassium chloride (KCl), these monovalent cations help stabilize the primer-template hybrid and optimize the chemical environment.
• Nuclease-Free Water: High-quality sterile water is used to bring the reaction to its final volume while ensuring the mixture is free from contaminating DNA and nucleases that could degrade the reaction.
Specialized Enzymes
The most critical enzyme in PCR is DNA polymerase, which must be heat-stable to survive the repeated high-temperature denaturation steps.
• Taq DNA Polymerase: The most commonly used enzyme, originally isolated from the thermophilic bacterium Thermus aquaticus found in hot springs. It is efficient but lacks proofreading activity, making it slightly error-prone.
• Pfu DNA Polymerase: Isolated from Pyrococcus furiosus, this enzyme is used when high accuracy is required because it possesses 3′ to 5′ proofreading ability.
• Reverse Transcriptase: This specialized enzyme is used in RT-PCR to convert RNA into complementary DNA (cDNA) before the standard PCR amplification begins.
• Uracil-N-Glycosylase (UNG): Used for contamination control, this enzyme excises uracil from previous reaction products (where dUTP was used instead of dTTP), preventing them from acting as templates in new reactions.
• Hot-Start Polymerases: These are modified versions of Taq that are inactivated at room temperature (often by an antibody or chemical inhibitor) and only become active after an initial heat activation step, reducing non-specific amplification.
Common Additives
For difficult templates, such as those with high GC content (>60%), researchers may add reagents like DMSO, formamide, or betaine to disrupt secondary structures and lower the melting temperature. Bovine Serum Albumin (BSA) is sometimes added to help neutralize inhibitors found in environmental or clinical samples
2. How do Pfu and Taq DNA polymerases differ in accuracy?
Pfu and Taq DNA polymerases differ primarily in their fidelity, or accuracy, during DNA synthesis. Taq DNA polymerase, originally isolated from Thermus aquaticus, is efficient and heat-stable but lacks proofreading activity (3′-5′ exonuclease activity). This absence of a correction mechanism makes Taq error-prone, as it can misincorporate nucleotides into the newly synthesized DNA strand.
In contrast, Pfu DNA polymerase, isolated from Pyrococcus furiosus, is widely used when high accuracy is required because it possesses a 3′ to 5′ proofreading ability. This function allows the enzyme to recognize and correct misincorporated nucleotides during the amplification process, resulting in significantly lower error rates than Taq.
According to the sources, there are specific trade-offs and applications for each:
• Application: Pfu is recommended for high-fidelity tasks like cloning and primer extension, where any sequence error is undesirable. Taq may be preferred when speed, yield, or processivity is more important than absolute accuracy.
• Speed: While more accurate, Pfu typically runs slower than Taq.
• Cost: Pfu is noted as being very expensive compared to Taq.
• Hybrid Solutions: To combine the benefits of both enzymes, researchers sometimes add a small amount of Pfu to a Taq reaction to improve accuracy while maintaining faster speeds.
While Taq provided a major advancement over the original E. coli polymerase by offering higher accuracy and heat stability, modern high-fidelity enzymes like Pfu (and others such as Q5) are now utilized to minimize mutations in amplified fragments
3. How does magnesium concentration affect the success of PCR?
Magnesium ions (Mg2+), typically added as magnesium chloride (MgCl2), act as an essential cofactor for thermostable DNA polymerases. The concentration of magnesium has a significant impact on PCR stringency, affecting both the yield and the precision of the reaction.
Magnesium concentration affects the success of PCR in the following ways:
• Yield vs. Specificity and Fidelity: Generally, increasing the concentration of Mg2+ will increase the total yield of the PCR product. However, higher concentrations decrease the specificity and fidelity of the DNA polymerase, making errors more likely during synthesis.
• Effects of Low Concentration: If magnesium levels are too low, the reaction will not proceed, resulting in a complete lack of PCR product.
• Effects of Excessive Concentration: Too much magnesium can prevent complete denaturation of the DNA template by over-stabilizing the double-stranded duplex. It also stabilizes spurious (incorrect) annealing of primers to non-target sites, which leads to the formation of undesired non-specific products, multiple bands, or a “smear” on an agarose gel.
• Interaction with dNTPs: Magnesium and deoxynucleoside triphosphates (dNTPs) have stoichiometric interactions. Because dNTPs bind to magnesium, higher concentrations of dNTPs in a reaction mixture reduce the amount of free magnesium available for the enzyme, necessitated an increase in MgCl2 concentration to compensate.
• Optimization and Titration: While many manufacturers provide buffers with a standard concentration of 1.5 mM MgCl2, many reactions require optimization through titration. Case studies show that some specific targets, such as the yeast GAL3 gene or certain mycobacteriophages, may require concentrations as high as 4.0 mM to produce a discrete, visible band.
Because changing the magnesium concentration can influence other parameters like annealing temperature, it is considered one of the most critical variables to manipulate when troubleshooting a failed or non-specific PCR experiment
4. Which PCR applications strictly require 3′ to 5′ proofreading activity?
In PCR, 3′ to 5′ proofreading activity is essential for high-fidelity applications where the introduction of mutations by error-prone enzymes like Taq polymerase would be unacceptable. This specialized enzymatic function is required for the following specific applications:
• Cloning: Accuracy is critical in cloning because PCR amplicons containing sequence errors are undesirable for downstream use. High-fidelity enzymes like Pfu or UITma DNA polymerase are utilized to ensure the amplified product is a perfect replica of the target DNA before it is inserted into a vector.
• DNA Sequencing: High-fidelity amplification is necessary for sequencing to guarantee that the final data accurately reflects the original template sequence.
• Primer Extension Reactions: Specialized primer extension tasks that require high-fidelity results rely on proofreading enzymes to maintain sequence integrity.
• Production of Error-Free Products: In any research or diagnostic setting where error-free amplicons are strictly required, standard Taq polymerase is bypassed in favor of modified polymerases with 3′-5′ exonuclease (proofreading) capabilities.
While standard Taq polymerase is efficient and widely used, its lack of proofreading activity makes it prone to misincorporating nucleotides, which can lead to a heterogeneous population of PCR products. Therefore, researchers use enzymes like Pfu polymerase for these specific tasks because its ability to recognize and correct errors results in significantly higher accuracy.
When should additives like DMSO or BSA be used?
Additives are specialized reagents used to optimize PCR when standard reaction conditions fail or produce suboptimal results, such as non-specific products or a lack of yield. The sources specify several conditions under which Dimethylsulfoxide (DMSO) and Bovine Serum Albumin (BSA) should be utilized.
When to Use Dimethylsulfoxide (DMSO)
DMSO is primarily used as an enhancer for difficult DNA templates and to improve primer specificity. It should be used in the following scenarios:
• GC-Rich Templates: When working with DNA that has a GC content greater than 60%, DMSO is added at a final concentration of 1–10%. It works by disrupting base pairing, which effectively lowers the melting temperature (Tm) and aids in the separation of these stable strands.
• Long PCR Products: DMSO, often in conjunction with glycerol and reduced potassium salt (KCl), is used to facilitate the amplification of longer fragments ranging from 10 to 40 kb.
• Resolving Primer Dimers: If primers preferentially self-anneal or anneal to each other rather than the template, adding DMSO can sometimes resolve this issue.
• Extremely GC-Rich Sequences: In “slowdown PCR,” which is designed for templates with GC content above 83%, DMSO is frequently used alongside other additives like Betaine.
Constraint Note: While beneficial, adding more than 2% DMSO can inhibit the activity of Taq DNA polymerase, potentially requiring the addition of more enzyme to the reaction.
When to Use Bovine Serum Albumin (BSA)
BSA is a specific protein additive used primarily to overcome the effects of PCR inhibitors found in biological or environmental samples. It should be used when:
• Analyzing Old or Degraded DNA: BSA helps alleviate problems associated with very old DNA samples, which frequently accumulate inhibitors over time.
• Neutralizing Specific Inhibitors: It is effective at aiding PCR in the presence of inhibitors such as FeCl3, hemin, fulvic acid, humic acid, and tannic acids.
• Difficult Environmental/Clinical Samples: BSA is used when working with DNA extracted from feces, fresh water, or marine water to neutralize contaminants that might otherwise lead to false-negative results.
Constraint Note: BSA does not alleviate inhibition caused by certain reagents, including EDTA, NaCl, SDS, or Triton X-100.
Other Related Additives
The sources also mention other additives for similar challenges:
• Betaine: Used for G-C rich targets, often in combination with DMSO, to reduce the dependence of DNA melting temperature on nucleotide composition.
• Formamide: Used like DMSO to disrupt base pairing, increase stringency, and enhance the amplification of GC-rich templates.
• Non-ionic Detergents (e.g., Triton X-100, Tween 20): Used at concentrations of 0.1 to 1% to suppress secondary structures and stabilize the DNA polymerase.
5. In what ways has PCR revolutionized medical diagnostics, forensic science, and genetic research?
The development of Polymerase Chain Reaction (PCR) has transformed biological science by allowing researchers to “pick the piece of DNA you’re interested in and have as much of it as you want” from trace amounts of material. This “molecular photocopying” technique has become the gold standard for specificity and sensitivity across several fields.
Medical Diagnostics
PCR has fundamentally altered how diseases are detected, monitored, and treated:
• Infectious Disease Detection: PCR allows for the rapid and highly specific diagnosis of bacterial and viral infections, including HIV, tuberculosis, COVID-19, and influenza. In the case of HIV, PCR can detect a single viral genome among 50,000 host cells, allowing for earlier detection, screening of donated blood, and precise monitoring of viral load to gauge treatment efficacy.
• Oncology: The technique permits the early diagnosis of malignant diseases like leukemia and lymphomas with a sensitivity 10,000 times higher than other methods. It also enables personalized healthcare through “liquid biopsies,” which identify specific mutations (e.g., EGFR or KRAS) in tumors to guide targeted drug therapies.
• Prenatal and Genetic Testing: PCR is used to diagnose hereditary disorders like cystic fibrosis and Huntington’s disease. It has revolutionized obstetric care through noninvasive prenatal diagnostics, which analyze fetal DNA circulating in maternal plasma, and preimplantation genetic diagnosis, where individual cells from an embryo are tested for mutations before IVF implantation.
• Tissue Typing: PCR is vital for tissue typing in organ transplantation to ensure compatibility.
Forensic Science
PCR-based DNA profiling (or fingerprinting) has become an indispensable analytical tool in criminal justice:
• Analysis of Minute Samples: Because PCR can amplify DNA from a single cell, investigators can generate conclusive evidence from a single hair root, a skin sample under a fingernail, or a spot of blood smaller than a pinhead.
• Identification and Exoneration: Forensic labs use PCR to match crime scene evidence to suspects or to the CODIS DNA database. This has been used to confirm the identity of perpetrators in decades-old “cold cases” and to exonerate individuals who were originally wrongly convicted.
• Paternity and Identification: The technique is the reliable standard for paternity testing and identifying human remains in missing persons cases. It can even be used for real-time sex determination from ancient or forensic bone samples.
Genetic Research
PCR has accelerated discovery by providing the high quantities of pure DNA necessary for molecular analysis:
• The Human Genome Project (HGP): PCR was a fundamental technology behind the early stages of the HGP, serving as a vital tool for mapping and sequencing the entire human set of genes.
• Acellular Cloning: PCR allows for “acellular cloning,” which produces millions of copies of a DNA fragment in hours—a process that previously took several days using bacteria and plasmid vectors.
• Gene Expression Analysis: Through RT-qPCR, researchers can measure gene expression levels in real-time to determine which genes are active or “switched on” in specific tissues or single cells.
• Evolutionary Biology and Paleogenetics: PCR has revolutionized the study of ancient DNA, allowing scientists to reconstruct the genomes of extinct species (such as mammoths) or analyze remains from Egyptian mummies and Neanderthals to understand phylogenetic relationships.
• Site-Directed Mutagenesis: Scientists use PCR to intentionally generate mutations in genes to study how specific proteins function or to improve their activity.
6. How does PCR differ for RNA versus DNA samples?
PCR for RNA samples differs from DNA samples primarily by requiring an additional initial step known as reverse transcription before the standard amplification cycles can begin. While standard PCR works directly on DNA templates, it cannot be performed on RNA without first converting the genetic information into a compatible form.
The key differences include:
• Enzymatic Conversion: In RNA-based PCR, a specialized enzyme called reverse transcriptase is used to read the RNA sequence and synthesize a complementary DNA (cDNA) copy. DNA-based PCR does not require this enzyme and uses DNA polymerase from the start.
• Template Nature: DNA is a double-stranded molecule that serves as a direct template for DNA polymerase after heat denaturation. RNA is typically single-stranded and must be transformed into double-stranded cDNA to undergo the standard cycles of denaturation, annealing, and extension.
• Stability: RNA is noted as being unstable, and the RT-PCR process effectively converts these unstable sequences into stable cDNA for analysis.
• Targeting and Primers: For RNA, primers can be designed to be complementary to specific RNA sequences or to the poly(A) tail (for messenger RNA) to begin the production of cDNA. In genomic DNA PCR, primers flank a specific region of the genome to be amplified.
• Content of Product: When PCR is performed on messenger RNA (mRNA), the resulting cDNA reflects only the coding sequences (exons) of a gene, whereas PCR performed on genomic DNA includes both exons and non-coding introns.
Applications of RT-PCR
This modified technique is essential for specific fields where the target genetic material is not DNA:
• Infectious Disease: It is used to diagnose infections caused by RNA viruses, such as SARS-CoV-2 (COVID-19), HIV, and influenza.
• Gene Expression: Researchers use RT-PCR to measure mRNA levels, which indicates which genes are active or “switched on” in a cell at a specific time.
• Oncology: Combined with quantitative methods (RT-qPCR), it allows for the sensitive detection of gene regulation and mutations associated with cancer.
7. How do GC-rich templates affect PCR efficiency?
GC-rich templates significantly reduce PCR efficiency because G-C base pairs contain three hydrogen bonds, whereas A-T pairs only have two. This increased chemical stability means that DNA with a high GC content requires higher melting temperatures and often longer times to achieve complete denaturation. Standard enzymes like Taq DNA polymerase are relatively unstable above 90°C, creating a challenge when the template requires extreme temperatures to separate the strands.
The impact of high GC content on PCR efficiency includes several specific technical hurdles:
• Secondary Structures: GC-rich sequences are highly prone to forming stable secondary structures, such as hairpin loops, in both the template and the resulting amplicons. These structures can prevent primers from binding to the single-stranded template or block the DNA polymerase from proceeding during the extension phase.
• Primer Design Challenges: Ideal primers should have a GC content between 40% and 60% to ensure stable annealing. In GC-rich regions, it is difficult to design primers that avoid mispriming, self-dimers, or hairpins, all of which can result in no product or non-specific amplification.
• Refractory Nature: Templates with a GC content greater than 60% are considered “refractory” and pose some of the greatest challenges to successful PCR.
To maintain efficiency with these difficult templates, researchers employ specialized additives and protocol modifications:
• Chemical Enhancers: Reagents like DMSO and Formamide are used to disrupt base pairing, which effectively lowers the melting temperature (Tm) and increases stringency. Betaine is another common additive that reduces the Tm dependence on nucleotide composition, aiding the amplification of GC-rich targets.
• Nucleotide Analogs: The analog dc7GTP (7-deaza-2′-deoxyguanosine 5′-triphosphate) can be used to destabilize secondary structures within the DNA product.
• Specialized Protocols: For sequences with extreme GC content (above 83%), a technique called “Slowdown PCR” is used. This method utilizes modern thermal cyclers to adjust ramp speeds and cooling rates, often in combination with dc7GTP, to ensure successful amplification.
8. How does DMSO lower DNA melting temperatures?
Dimethylsulfoxide (DMSO) lowers DNA melting temperatures (Tm) by disrupting the hydrogen bonds involved in base pairing between complementary DNA strands. By chemically weakening these interactions, DMSO destabilizes the double helix, making it easier for the strands to separate into single-stranded templates at lower temperatures.
This mechanism is particularly important for the following reasons:
• GC-Rich Templates: DNA with a GC content greater than 60% is naturally more stable because G-C base pairs share three hydrogen bonds, whereas A-T pairs only share two. DMSO’s ability to disrupt these bonds effectively lowers the Tm, facilitating the amplification of these “refractory” or difficult sequences.
• Enzyme Protection: Standard DNA polymerases like Taq can become unstable or inactivated at temperatures above 90°C. By lowering the Tm of the template, DMSO allows complete denaturation to occur at temperatures that do not destroy the enzyme’s activity.
• Secondary Structures: DMSO helps prevent the formation of, or helps disrupt, stable secondary structures like hairpin loops in the template or amplicons, which can otherwise block the progress of the DNA polymerase.
Usage Constraints: DMSO is typically used in PCR at a final concentration of 1% to 10%. However, researchers must be cautious because concentrations higher than 2% can inhibit Taq DNA polymerase, which may require the addition of more enzyme to the reaction to compensate. DMSO is also frequently used in combination with other additives like Betaine to further reduce the dependence of the melting temperature on the specific nucleotide composition of the DNA.
9. What other inhibitors can BSA help neutralize?
Bovine Serum Albumin (BSA) is a protein additive used in PCR primarily to neutralize a variety of biological and environmental inhibitors that can interfere with the activity of DNA polymerase. According to the sources, BSA is specifically effective at aiding PCR in the presence of the following inhibitors:
• Organic Acids: It helps mitigate the effects of humic acid, fulvic acid, and tannic acids, which are common in environmental samples.
• Metallic and Heme Compounds: BSA can neutralize ferric chloride (FeCl3) and hemin.
• Contaminated Sample Extracts: It is used to facilitate successful amplification from extracts of feces, fresh water, and marine water.
• Degradation Products in Old Samples: The addition of BSA can help alleviate problems with very old DNA, which often accumulates inhibitors over time.
While highly effective against the substances listed above, the sources specify that BSA cannot neutralize every type of inhibitor; it does not provide relief against bile salts, bilirubin, EDTA, NaCl, SDS, or Triton X-100. Additionally, in some cases, specialized proteins like T4 gene 32 protein may be used alongside or in place of BSA to achieve similar neutralizing effects.
10. How do GC-rich templates affect PCR efficiency?
GC-rich templates (typically defined as those with a GC content greater than 60%) significantly reduce PCR efficiency and are considered some of the greatest challenges in the procedure. This reduction in efficiency is caused by the chemical stability of the G-C base pair, which contains three hydrogen bonds, whereas A-T pairs only have two.
The impact of high GC content on PCR efficiency includes several specific technical hurdles:
1. Resistance to Denaturation
Because G-C bonds are stronger than A-T bonds, GC-rich DNA requires higher melting temperatures for complete strand separation. This is problematic because standard enzymes like Taq DNA polymerase are relatively unstable above 90°C. If the temperature required to denature the template is too high, the polymerase may lose its enzymatic activity before amplification can succeed.
2. Formation of Secondary Structures
GC-rich templates and their resulting amplicons are highly prone to forming stable secondary structures, such as hairpin loops. These structures can:
• Prevent primers from binding to the single-stranded template.
• Block DNA polymerase from proceeding along the template during the extension phase, leading to incomplete products.
3. Primer Design and Specificity
While an optimal GC content for primers is between 40% and 60% to ensure stable annealing, templates with extreme GC richness make it difficult to design specific primers. In these regions, primers are more likely to undergo mispriming, form self-dimers, or create hairpins, all of which decrease the yield of the desired product.
4. Overcoming GC-Rich Challenges
To maintain efficiency with these difficult templates, researchers use specialized protocol modifications and additive reagents:
• Chemical Enhancers: DMSO (1–10%) and Formamide are used to disrupt base pairing, which effectively lowers the melting temperature (Tm) and aids strand separation. Betaine is also used to reduce the Tm dependence on nucleotide composition.
• Nucleotide Analogs: The analog dc7GTP can be incorporated into the reaction to destabilize secondary structures in the product.
• Specialized Protocols: For sequences with extreme GC content (above 83%), a technique called “Slowdown PCR” is employed. This method utilizes modern thermal cyclers to adjust ramp speeds and cooling rates to ensure successful annealing and extension despite the stable nature of the template.
11. How do dNTPs and magnesium ions interact during the reaction?
Deoxynucleoside triphosphates (dNTPs) and magnesium ions (Mg2+) have a stoichiometric interaction in which dNTPs bind to magnesium. This interaction effectively reduces the amount of free magnesium available in the reaction mixture.
Because thermostable DNA polymerases strictly require magnesium to act as a cofactor for their enzymatic activity, the reaction cannot proceed if the level of free ions is insufficient. The relationship between these two components results in several practical considerations for PCR optimization:
• Compensatory Adjustments: A large increase in the dNTP concentration will necessitate a corresponding increase in the concentration of magnesium (MgCl2) to compensate for the binding and maintain enough free cofactor for the enzyme.
• Enzyme Function: If dNTPs sequester too much magnesium, leaving the concentration of free ions too low, the reaction will fail to proceed, resulting in a lack of PCR product.
• Reaction Yield vs. Specificity: While increasing magnesium can improve the yield of a reaction that is struggling due to dNTP consumption, excessive magnesium can decrease the specificity and fidelity of the DNA polymerase.
• Standard Formulations: Most standard PCR buffers are designed with approximately 1.5 mM MgCl2 to account for typical dNTP concentrations, but titration is often required if reagent concentrations are altered.
13. What happens if the magnesium concentration is set too low?
If the magnesium concentration (Mg2+) is set too low, the PCR reaction will not proceed, ultimately resulting in no PCR product. This occurs because magnesium ions act as an essential cofactor that thermostable DNA polymerases strictly require to catalyze the synthesis of new DNA strands.
The consequences of insufficient magnesium include:
• Enzyme Inactivity: Because the enzyme requires these ions to function, the reaction cannot initiate or continue if they are missing or present in inadequate amounts.
• Lack of Amplicons: In experimental trials, such as those involving the yeast GAL3 gene or mycobacteriophages, researchers observed that concentrations below a specific threshold resulted in no visible bands on an agarose gel.
• Sensitivity to dNTP Levels: Even if magnesium is added to the reaction, the concentration may be effectively “too low” if dNTP levels are high; this is because dNTPs bind to magnesium, reducing the amount of free cofactor available for the DNA polymerase.
• Troubleshooting Failure: A total lack of product is one of the most common indicators that the magnesium concentration needs to be titrated or increased for a specific set of primers or templates.
14. Why does high magnesium lead to non-specific PCR products?
High concentrations of magnesium (Mg2+) lead to non-specific PCR products primarily because they reduce the stringency of the reaction and stabilize incorrect primer-template interactions.
The specific mechanisms by which excessive magnesium compromises PCR specificity include:
• Stabilization of Spurious Annealing: Too much magnesium stabilizes the annealing of primers to non-target sites on the DNA template that are only partially complementary. This allows the DNA polymerase to initiate synthesis from these incorrect locations, resulting in undesired PCR products often visualized as “multiple bands” or a “smear” on an agarose gel.
• Reduced Fidelity: Increased magnesium levels decrease the fidelity of the DNA polymerase enzyme. This makes the enzyme more likely to misincorporate incorrect nucleotides during synthesis, which can lead to mutations or heterogeneous product populations.
• Incomplete Denaturation: Excessive magnesium can prevent the complete denaturation of the DNA template by over-stabilizing the double-stranded duplex. If the strands do not separate fully during the heating phase, it interferes with correct primer binding and efficient amplification.
• Enhanced Enzyme Activity at Incorrect Sites: As an essential cofactor, magnesium is required for the polymerase to function; however, an overabundance can drive the enzyme to extend poorly matched primer-template hybrids that would otherwise remain unstable under more stringent conditions.
Because of these effects, titrating the magnesium concentration (typically provided as MgCl2) is a critical step in PCR optimization to balance a high yield of the desired product with the necessary specificity to avoid non-specific amplification.
15. How do dNTP concentrations influence overall PCR yield and precision?
Deoxynucleoside triphosphates (dNTPs) serve as the essential building blocks and provide the energy required for the DNA polymerase to synthesize new DNA strands. Their concentration is a critical variable that must be balanced to ensure both high product yield and sequence precision.
Influence on PCR Yield
The availability of dNTPs directly dictates the quantity of DNA that can be produced:
• Reagent Limitation and the Plateau Effect: As the PCR progresses, the consumption of dNTPs causes them to become a limiting factor. This depletion, along with the accumulation of pyrophosphate and the inactivation of the enzyme, leads to a “plateau effect” where the reaction ceases to amplify at an exponential rate.
• Minimum Requirements: If dNTPs are inadequate or old (degraded by repeated freezing and thawing), the reaction will fail to proceed, resulting in no detectable PCR product.
• Fragment Length: While the standard final concentration is typically 200 μM (50 μM of each of the four nucleotides), higher concentrations may be required when amplifying longer PCR fragments.
• Inhibition by Excess: While necessary for yield, excessive dNTP concentrations can actually inhibit the PCR process.
Influence on Precision (Specificity and Fidelity)
The concentration of dNTPs significantly impacts the accuracy and stringency of the reaction:
• Higher Accuracy at Lower Levels: Utilizing lower concentrations of dNTPs is a known strategy to increase both the specificity and fidelity of the reaction. This reduces the likelihood of the DNA polymerase misincorporating incorrect nucleotides or extending non-specific primer-template matches.
• Equivalent Concentrations: Problems can arise if the four types of dNTPs (A, T, C, and G) are not maintained at equivalent concentrations; an imbalance can compromise the precision of the synthesis.
• Stoichiometric Interaction with Magnesium: There is a critical relationship between dNTPs and magnesium ions. Because dNTPs bind to magnesium, high concentrations of dNTPs reduce the amount of free magnesium available to act as a cofactor for the DNA polymerase. Consequently, a large increase in dNTPs necessitates a corresponding increase in magnesium concentration (MgCl2) to maintain enzymatic activity, which can further influence the overall stringency of the reaction.
Specialized dNTP Variants
Beyond standard dNTPs, specialized versions can be used to improve precision or prevent errors:
• dUTP: Substituting dUTP for dTTP creates uracil-containing amplicons, which can be treated with the enzyme uracil-N-glycosylase (UNG) to prevent carryover contamination from previous reactions.
• 7-deaza-2′-deoxyguanosine (dc7GTP): This analog is used to destabilize secondary structures in the DNA product, facilitating the amplification of difficult, GC-rich templates.
16. Are there other cofactors besides magnesium that enzymes might require?
Enzymes used in PCR, particularly DNA polymerase, primarily require magnesium (Mg2+) as an essential cofactor, but they can also utilize manganese (Mn2+) ions. While magnesium is the most common bivalent cation used to achieve optimal results, manganese can be substituted for specialized applications such as PCR-mediated DNA mutagenesis. This substitution is beneficial for specific research goals because a higher concentration of manganese increases the error rate during the synthesis of new DNA strands.
Beyond these bivalent cations, PCR reactions typically require monovalent cations, most frequently potassium (K+) ions. These ions are usually provided in the form of potassium chloride (KCl) and are necessary to maintain a suitable chemical environment for the optimum activity and stability of the DNA polymerase.
While the sources focus on the ionic requirements for DNA polymerase, other specialized enzymes mentioned in the context of PCR—such as reverse transcriptase for RNA conversion or uracil-N-glycosylase (UNG) for contamination control—also operate within these buffered environments containing these salts, though their specific metal cofactor requirements are not detailed in the provided materials.
17. What happens to the DNA polymerase enzyme if magnesium is too high?
When magnesium (Mg2+) concentrations are too high, the most significant impact on the DNA polymerase enzyme is a decrease in its specificity and fidelity.
This excessive concentration affects the enzyme and the reaction in the following specific ways:
• Reduced Fidelity: The DNA polymerase becomes more prone to errors, meaning it is more likely to misincorporate incorrect nucleotides during the synthesis of new DNA strands.
• Stabilization of Spurious Annealing: High levels of magnesium stabilize the “spurious” (incorrect) annealing of primers to non-target sites on the DNA template that are not perfectly complementary.
• Non-Specific Amplification: Because the enzyme remains active but less precise, it will extend these incorrectly bound primers, resulting in the production of undesired non-specific PCR products. On an agarose gel, this is typically visualized as multiple bands or a “smear”.
• Inhibition of Denaturation: Too much magnesium can over-stabilize the double-stranded DNA duplex, which may prevent complete denaturation during the heating phase. If the template does not separate into single strands, the DNA polymerase cannot initiate synthesis.
While magnesium is an essential cofactor for thermostable DNA polymerases, an unoptimized, high concentration reduces the stringency of the reaction, leading to excess non-specific products rather than a clean, singular amplicon.
18. How does dNTP depletion lead to the plateau effect?
DNTPs (deoxynucleoside triphosphates) are the essential building blocks that DNA polymerase uses to synthesize new DNA strands during the extension phase of each PCR cycle,. As the reaction progresses through repeated cycles, these nucleotides are individually incorporated into the growing DNA chains, leading to their gradual consumption,.
DNTP depletion leads to the plateau effect through the following mechanisms:
• Substrate Limitation: The total amount of amplified product is determined by the available substrates; as dNTPs are consumed, they eventually become a limiting factor that slows the reaction rate,.
• Loss of Exponential Growth: During the early cycles, DNA copies double every round (exponential amplification). However, once dNTP concentrations become insufficient to support this rate, the reaction enters a leveling-off stage where it can no longer amplify the target sequence at an exponential rate,.
• Reaction Cessation: When dNTPs reach a point of exhaustion, the DNA polymerase can no longer synthesize new strands, and no more product accumulates, resulting in the final plateau stage,.
While dNTP depletion is a primary driver, the plateau effect is also influenced by other factors, including the loss of DNA polymerase activity, the consumption of primers, and the inhibitory effect of accumulated amplicons (the reaction products),,. Because this effect makes end-point quantification unreliable, researchers often turn to Real-Time Quantitative PCR (qPCR) to analyze the reaction during its earlier exponential phase.
19. How is the ideal magnesium concentration determined for new samples?
The ideal magnesium concentration for a new sample is determined through a process called titration, where multiple reactions are run simultaneously with varying amounts of magnesium to identify which concentration produces the best result., While many manufacturers provide a standard 10X buffer that results in a final concentration of 1.5 mM MgCl2, this amount is often insufficient for new or uncharacterized templates.,
According to the sources, the following steps are used to determine the optimal concentration:
• Establish a Testing Range: Researchers typically test a concentration range from 0.0 mM to 5.0 mM.,
• Use Incremental Steps: The titration is usually performed in set increments (e.g., 0.5 mM steps) to find the precise balance between yield and specificity.,
• Maintain Constant Variables: When troubleshooting or optimizing for a new sample, only one reagent should be manipulated at a time; therefore, all other components like primers, dNTPs, and template DNA must remain at constant concentrations across all test tubes.
• Evaluate via Gel Electrophoresis: After the PCR is complete, the products are analyzed on an agarose gel. The “ideal” concentration is identified as the one that produces a discrete, visible band of the expected size while avoiding non-specific products, multiple bands, or a “smear.”,,
• Adjust for Reagent Interactions: If the protocol requires high concentrations of dNTPs, the magnesium concentration must be increased accordingly because dNTPs bind to magnesium, reducing the amount of free cofactor available for the DNA polymerase.,
Case studies in the sources demonstrate that for new samples, such as the yeast GAL3 gene or certain mycobacteriophages, the titration process often reveals an optimal concentration of 4.0 mM, which is significantly higher than the standard 1.5 mM recommendation., Conversely, if the magnesium level is set too low for a new sample, the reaction will fail to proceed entirely, resulting in no detectable PCR product.
20. How do you choose a starting magnesium concentration for optimization?
When choosing a starting magnesium concentration for PCR optimization, the standard practice is to begin with a final reaction concentration of 1.5 mM MgCl2. This specific starting point is widely adopted because many manufacturers provide 10X PCR buffer solutions that already contain 15 mM MgCl2, which translates to a 1.5 mM final concentration when diluted for a typical reaction.
While 1.5 mM is often sufficient for standard applications, the following factors should be considered when selecting or adjusting a starting point for optimization:
• Optimization Range: If the standard 1.5 mM concentration fails to produce a satisfactory result, researchers typically explore a range between 0.5 mM and 5.0 mM.
• Titration Increments: When attempting to find the ideal level for a new or uncharacterized sample, a common optimization strategy is to perform a titration using 0.5 mM increments.
• Target DNA Characteristics: For certain uncharacterized templates, such as mycobacteriophages or specific yeast genes, the standard 1.5 mM starting point may be entirely insufficient. Case studies show that for these “temperamental” targets, optimal amplicon production may not occur until the concentration reaches 4.0 mM.
• Stoichiometric Interaction with dNTPs: The starting magnesium concentration must be adjusted based on the dNTP concentration used in the reaction. Because dNTPs bind to magnesium ions, any protocol that utilizes high concentrations of dNTPs (such as for long-range PCR) will require a correspondingly higher starting concentration of magnesium to ensure enough free ions are available to act as a cofactor for the DNA polymerase.
• Balancing Yield and Specificity: When choosing a starting concentration, researchers must weigh the desire for high yield against the need for accuracy. Higher concentrations generally increase yield but decrease the specificity and fidelity of the DNA polymerase, which can lead to non-specific products or mutations. Conversely, if the starting concentration is too low, the reaction will not proceed at all.
21. How do you calculate the optimal annealing temperature for primers?
Calculating the optimal annealing temperature (Ta) is essential for ensuring that primers bind specifically to their target sequence with high efficiency. The process begins by determining the Melting Temperature (Tm) of the primers, which is the temperature at which half of the DNA duplexes are separated into single strands.
Standard Melting Temperature (Tm) Calculations
There are two primary methods for estimating the Tm of a primer:
• Simple (Wallace) Formula: For primers that are short (no longer than 25 nucleotides), the most common calculation used is: Tm=4(G+C)+2(A+T)∘C In this formula, G, C, A, and T represent the number of those specific nucleotides in the primer sequence.
• Nearest-Neighbor Thermodynamic Model: This is considered the most accurate method because it accounts for the stacking energy of neighboring base pairs. The calculation is more complex: Tm (Primer)=((ΔH/(ΔS+R×ln(c/4)))−273.15+16.6log[K+]) Where ΔH is the sum of nearest-neighbor enthalpy changes, ΔS is entropy changes, R is the Gas Constant, C is the primer concentration, and [K+] is the potassium concentration (typically 50 mM).
Calculating the Optimal Annealing Temperature (Ta)
Once the Tm is known, the optimal annealing temperature can be determined using several approaches:
• The Optimal Ta Equation: A more precise mathematical approach for any PCR reaction uses the following formula: Ta OPT=0.3×Tm Primer+0.7×Tm Product−14.9. In this equation, the Tm of the product (amplicon) is calculated as: Tm Product=0.41(%G−C)+16.6log[K+]−675/product length.
• General Rule of Thumb: In standard practice, the annealing temperature is typically set 3–5 ∘C below the calculated Tm of the primers. Some sources suggest a tighter range of 1–2 ∘C below the Tm to minimize mismatches.
Experimental and Alternative Methods
If mathematical calculations fail to produce a discrete band, researchers use the following strategies:
• Gradient Thermal Cyclers: These allow for the experimental determination of the best temperature by running the same reaction at slightly different temperatures across multiple samples simultaneously.
• Touchdown PCR: This technique bypasses precise calculation limitations by starting at a high annealing temperature (e.g., 10 ∘C above the Tm) and gradually lowering it by 1 ∘C every cycle or every other cycle, which increases specificity.
• Primer Matching: It is critical that the two primers in a set have similar Tm values—ideally differing by no more than 5 ∘C—so they can both bond with equal ease at the chosen annealing temperature.
22. How do primer melting temperatures impact the overall PCR success?
The primer melting temperature (Tm)—defined as the temperature at which half of the DNA duplexes are separated into single strands—is a critical factor because it dictates the annealing temperature (Ta), which directly determines the specificity and yield of the PCR reaction.
Impact on Specificity and Yield
The success of PCR relies on setting an annealing temperature relative to the primers’ Tm. The sources outline two primary risks of incorrect temperature settings:
• Temperature Too High: If the annealing temperature is set too high (typically above the Tm), the primers may fail to bind to the template DNA at all, resulting in no PCR product.
• Temperature Too Low: If the temperature is too low, the hybridization becomes less selective. This allows primers to bind imperfectly to non-target sequences, leading to the amplification of non-specific products, visualized as multiple bands or a “smear” on an agarose gel.
In standard practice, the optimal annealing temperature is typically set 3–5°C below the Tm of the primers to ensure stable and specific binding.
Coordination Between Primer Pairs
For a PCR reaction to be successful, the two primers used (forward and reverse) must have similar Tm values. According to the sources:
• The Tm of both primers should ideally differ by no more than 5°C.
• If the Tm values are drastically different, it becomes difficult to select a single annealing temperature that allows both primers to bind to their targets with high efficiency.
• Designing primers with similar Tm values ensures that both can bond with equal ease under the same thermal cycling conditions.
Factors Influencing Tm
Several variables affect the melting temperature and, consequently, the reaction’s success:
• GC Content: G-C base pairs share three hydrogen bonds, while A-T pairs only share two. Therefore, primers with a higher GC content have a higher Tm and require more energy to denature. The optimal GC content for primers is generally 40–60%.
• Primer Length: Optimal primers are usually 18–22 base pairs long. Length influences the rate of hybridization; if a primer is too long, the efficiency of the PCR may be reduced.
• Additives: Reagents like DMSO can be used to disrupt base pairing, effectively lowering the Tm to help amplify difficult or GC-rich templates.
Optimization Strategies
Because Tm calculations are only estimates, researchers use specific techniques to find the best temperature for success. Touchdown PCR starts with an annealing temperature approximately 10°C above the calculated Tm and gradually lowers it each cycle, which confers a significant advantage to the correct product over non-specific ones. Additionally, gradient thermal cyclers allow for the experimental testing of various temperatures in a single run to identify the one that produces the cleanest, most discrete band.
23. What happens to the DNA copies during the final hold step?
During the final hold step, the thermal cycler cools the reaction chamber to a temperature typically ranging from 4°C to 15°C. This step occurs immediately after the final elongation phase is complete.
The primary functions of the final hold include:
• Short-Term Storage: The low temperature serves to preserve the amplified DNA products (amplicons) for a brief period. It acts as a holding phase that keeps the DNA stable until the researcher can remove the tubes from the machine.
• Reaction Termination: By chilling the mixture, the hold step effectively stops the activity of the DNA polymerase, terminating the biochemical reaction.
• Indefinite Maintenance: Most modern thermal cyclers are designed to maintain this 4°C temperature indefinitely. This allows the machine to run overnight or while the researcher is away from the laboratory without risking the degradation of the samples.
• Preparation for Analysis: The hold keeps the DNA intact and ready for downstream applications, such as gel electrophoresis, DNA sequencing, or molecular cloning.
It is important to distinguish this from the final elongation step, which occurs just before the hold at a higher temperature (roughly 70–74 °C) to ensure that any remaining single-stranded DNA is fully extended and completed.
24. What happens to the DNA copies during the final hold step?
During the final hold step, the thermal cycler cools the reaction chamber to a temperature typically ranging from 4°C to 15°C. This step occurs immediately after the final elongation phase is complete.
The primary functions of the final hold include:
• Short-Term Storage: The low temperature serves to preserve the amplified DNA products (amplicons) for a brief period. It acts as a holding phase that keeps the DNA stable until the researcher can remove the tubes from the machine.
• Reaction Termination: By chilling the mixture, the hold step effectively stops the activity of the DNA polymerase, terminating the biochemical reaction.
• Indefinite Maintenance: Most modern thermal cyclers are designed to maintain this 4°C temperature indefinitely. This allows the machine to run overnight or while the researcher is away from the laboratory without risking the degradation of the samples.
• Preparation for Analysis: The hold keeps the DNA intact and ready for downstream applications, such as gel electrophoresis, DNA sequencing, or molecular cloning.
It is important to distinguish this from the final elongation step, which occurs just before the hold at a higher temperature (roughly 70–74 °C) to ensure that any remaining single-stranded DNA is fully extended and completed.
25. What causes DNA to degrade if not held at 4°C?
DNA is held at 4°C at the conclusion of a PCR run primarily to prevent nuclease activity, which can break down the genetic material. Nucleases are enzymes that may be present in the reaction mixture or contaminating the water, and they are capable of degrading both the DNA template and the newly synthesized amplicons.
According to the sources, the specific reasons for this cooling step include:
• Halting Enzymatic Activity: Chilling the mixture effectively stops the activity of the DNA polymerase, which terminates the biochemical reaction and prevents any further, potentially non-specific, synthesis.
• Preventing Smearing: If DNA is allowed to degrade, it often appears as a smear rather than a discrete band when analyzed via agarose gel electrophoresis.
• Preserving Stability: The low temperature serves as a short-term storage phase that keeps the amplified DNA products stable until they can be removed from the thermal cycler for analysis.
• Counteracting Fragmentation: Outside of controlled laboratory conditions, DNA naturally breaks down by fragmentation after the death of an organism; maintaining a cold, sterile environment in the PCR tube helps prevent this natural decay process.
To further ensure the integrity of the samples, researchers are advised to use nuclease-free water and keep all reagents on ice during the initial setup to minimize any degradation before the reaction even begins.
References
Analytical Methods Committee. (2014). PCR-the polymerase chain reaction. Analytical Methods, 6(2), 333–336. https://doi.org/10.1039/c3ay90101g
Garibyan, L., & Avashia, N. (2013). Polymerase chain reaction. Journal of Investigative Dermatology, 133(3), 1–4. https://doi.org/10.1038/jid.2013.1
Kadri, K. (2020). Polymerase Chain Reaction (PCR): Principle and Applications. In Synthetic Biology – New Interdisciplinary Science. IntechOpen. https://doi.org/10.5772/intechopen.86491
SL, L., & D, R. (2023). A Review of PCR, Principle, and Its Applications. International Journal of Pharmaceutical Research and Applications, 8(3), 3502. https://doi.org/10.35629/7781-080335023513
Lorenz, T. C. (2012). Polymerase chain reaction: Basic protocol plus troubleshooting and optimization strategies. Journal of Visualized Experiments, 63. https://doi.org/10.3791/3998
Suchman, E. (2011). Polymerase Chain Reaction Protocol. http://www.promega.com/biomath/calc11.htm#disc
https://en.wikipedia.org/wiki/History_of_polymerase_chain_reaction
https://en.wikipedia.org/wiki/Polymerase_chain_reaction
https://www.biochain.com/blog/the-polymerase-chain-reaction-what-it-is-and-how-it-works/
https://www.ncbi.nlm.nih.gov/probe/docs/techpcr/
https://www.genome.gov/about-genomics/fact-sheets/Polymerase-Chain-Reaction-Fact-Sheet
https://www.sciencelearn.org.nz/resources/2347-what-is-pcr
https://www.nature.com/scitable/definition/polymerase-chain-reaction-pcr-110/
https://www.thermofisher.com/np/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/pcr-education/pcr-reagents-enzymes/pcr-basics.html
https://learn.genetics.utah.edu/content/labs/pcr/
Related posts:
 Southern Blotting: A Comprehensive Guide to Molecular DNA Analysis and Protocol Principles
Southern Blotting: A Comprehensive Guide to Molecular DNA Analysis and Protocol Principles
 The Secret of the Blue and White Colonies: An Introduction to Blue-White Screening
The Secret of the Blue and White Colonies: An Introduction to Blue-White Screening
 The Genetic Engineer’s Toolkit: A Beginner’s Guide to Cloning Vectors
The Genetic Engineer’s Toolkit: A Beginner’s Guide to Cloning Vectors
 Molecular Scissors: An Introduction to Restriction Enzymes
Molecular Scissors: An Introduction to Restriction Enzymes
 Artificial Chromosomes: YACs, BACs, MACs, HACs
Artificial Chromosomes: YACs, BACs, MACs, HACs

Pingback: Comprehensive Technical Notes on Random Amplified Polymorphic DNA (RAPD) Analysis - Aneknowledge.com