
Contents
Introduction:
Cystic fibrosis (CF) is one of the most common autosomal recessive disorders affecting the respiratory system, digestive system, and other organs of the body1. Traditionally, the prevalence of CF was thought to be largely in Caucasians of North European origin with its predominance only in Europe, North America, and Australasia, however, recent studies have revealed its presence in other areas such as Latin America, Africa, and Middle East Asia at lower rates2. The epidemiology of CF varies widely according to geography and race/ethnicity. According to the Annual Data Report, 2019 of the Cystic Fibrosis Foundation, 31,000 American people were affected with CF in the US. Globally, approximately 79,000 people are living with CF with the highest prevalence in North America, Australia, and Europe3.
Pathogenesis:
It is caused due to a mutation in a single gene on chromosome 7 that encodes the cystic fibrosis transmembrane conductance regulator (CFTR) protein, hence the name given Cystic Fibrosis. The most predominant form of mutation is deletion, deleting the amino acid phenylalanine at position 508. It is called an autosomal recessive disease because individuals with only a single copy of the defective gene cannot cause the disease but make them carriers. CFTR proteins behave as chloride channels to regulate the flow of chloride ions across the cell membranes. These proteins maintain salt and water balance in the secreted fluids as chloride is responsible for driving the movement of water. Upon mutation of the gene, CFTR gets disrupted hence, deregulating the balance. Till now, more than 2,000 different variants of CFTR have been reported4.
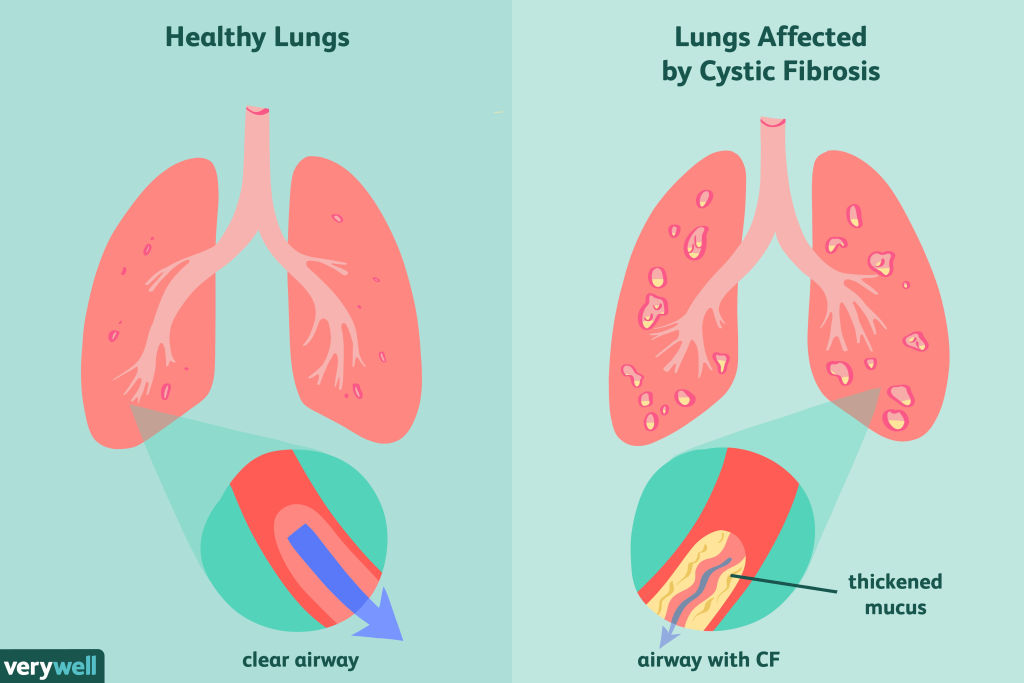
It is an inheritable disease that affects the cells producing mucus, sweat, digestive juice, and other bodily fluids. Typically, the fluids are thin and slippery and can easily pass through due to properly functioning CFTR proteins but in people with CF, the secreted fluids become thick and sticky as a consequence of mutated CFTR. Instead of flowing, they clog the ducts, tubes, and passageways, halting the function of organs, majorly, the pancreas and the lungs. When the air passageway gets blocked, people have difficulty breathing or cannot breathe at all. Similarly, the blockage in pancreatic ducts hinders the process of digestion1.
Symptoms:
The signs and symptoms of CF vary depending on the level of severity and the severity differs with the mutation type. The most severe form of CF is that which develops early in life. If the symptoms appear later, the severity of the disease decreases due to the prevalence of a milder CF. CF is a multi-system disorder, majorly affecting respiratory, digestive, and urogenital organs5.
Digestive symptoms:
Digestive problems manifest first than any other symptoms. The thick mucus obstructs bile ducts leading to poor absorption of fats, and increasing the probability of liver damage. Moreover, blockage of pancreatic ducts prevents the transport or synthesis of pancreatic enzymes into the small intestine. This results in poor digestion and accumulation of digestive enzymes or their precursors in the pancreas, destroying pancreatic tissues and leading to inflammation. Damage to the pancreas ultimately results in other complications like a shortage of beta-cells producing insulin, leading to diabetes mellitus.
The following symptoms can be observed6:
- Abdominal distension
- Intestinal obstruction
- Intussusception
- Loss of appetite
- Fatigue
- Nausea
- Slow weight gain and growth in children
- Severe constipation
Respiratory symptoms:
Thick mucus in the lungs becomes the barrier for air passageway preventing the flow of air in and out that causes breathing problems. The accumulation of mucus provides space for bacteria to grow and thrive leading to chronic infections.
Some of the common symptoms include7:
- Wheezing
- Consistent cough with thick mucus
- Recurrent pneumonia
- Exercise intolerance
- Stuffy nose
- Frequent lung infections
Urogenital symptoms:
CF can lead to infertility in men and decreased fertility in women8.
Some of the symptoms are:
- Delayed sexual development
- Irregular menstruation cycle
- Absence or halt of menstruation
- Cervix inflammation
Diagnosis:
Universal New-born screening (NBS):
It was introduced in the 1980s that allows CF screening of asymptomatic infants. It measures the amount of immunoreactive trypsinogen (IRT) in an infant’s blood. For this, a few drops of blood are pricked from the infant’s heel.
Trypsinogen is converted into a pancreatic enzyme called trypsin. In patients with CF, thick mucus blocks the pancreatic ducts and prevents the flow of trypsinogen into the small intestine, blocking the synthesis of trypsin. Thus, those with higher IRT levels are considered to show CF-positive results if the IRT levels (>60 ng/ml) remain elevated consistently between 7- 14 days of infancy. This test is then followed by genetic testing for further confirmation9.
CFTR Mutation Detection:
It is genetic testing done to detect the presence of CFTR mutation and possible CFTR variants. Typically, mutant variants are determined by identifying two known pathogenic variants on separate chromosomes. Initially, it is done to determine 100 mutations at a time. In most cases, a complete sequencing of the CFTR gene is recommended to confirm whether a person suffers from CF or not10.
Sweat test:
It is the gold standard for testing CF which measures the amount of chloride ions in an individual’s sweat. It should be performed after 48 hours of birth because within 24 hours infants have temporarily elevated sweat sodium levels. It is, generally a follow-up test done after a positive NBS result. For adequate sweat collection, infants should weigh more than 2 kg. If this shows an abnormal result, it should either be repeated another day or confirmed with genetic testing11.
References
1. Chen Q, Shen Y, Zheng J. A review of cystic fibrosis : Basic and clinical aspects. 2021;(August):220-232. doi:10.1002/ame2.12180
2. Guo J, Garratt A, Hill A. Worldwide rates of diagnosis and effective treatment for cystic fibrosis. J Cyst Fibros. 2022;21(3):456-462. doi:10.1016/j.jcf.2022.01.009
3. Leonard A, Bailey J, Bruce A, Jia S, Stein A, Fulton J, Helmick M, Litvin M, Patel A, Powers KE, Reid E, Sankararaman S, Clemm C, Reno K, Hempstead SE, DiMango E. Nutritional considerations for a new era: A CF foundation position paper. J Cyst Fibros. 2023 Sep;22(5):788-795. doi: 10.1016/j.jcf.2023.05.010. Epub 2023 May 23. PMID: 37230807.
4. Dickinson KM, Collaco JM. Cystic Fibrosis. 2023;(May). doi:10.1542/pir.2019-0212
5. Singireddy, S. R. S. R., Varagandhi, S., Jagiri, A. G., & Kadarla, R. K. (2019). An Overview on Cystic Fibrosis. Asian Journal of Pharmaceutical Research and Development, 7(5), 80-91.
6. Li L, Somerset S. Digestive system dysfunction in cystic fibrosis: challenges for nutrition therapy. Dig Liver Dis. 2014 Oct;46(10):865-74. doi: 10.1016/j.dld.2014.06.011. Epub 2014 Jul 19. PMID: 25053610.
7. Mccolley SA, Ren CL, Schechter MS, Regelmann WE, Pasta DJ, Konstan MW. Risk Factors for Onset of Persistent Respiratory Symptoms in Children With Cystic Fibrosis. 2011;(April):1-7. doi:10.1002/ppul.22519
8. Blau H, Freud E, Mussaffi H, Werner M, Konen O, Rathaus V. Urogenital abnormalities in male children with cystic fibrosis. Arch Dis Child. 2002 Aug;87(2):135-8. doi: 10.1136/adc.87.2.135. PMID: 12138064; PMCID: PMC1719175.
9. Scotet V, Gutierrez H, Farrell PM. Newborn Screening for CF across the Globe-Where Is It Worthwhile? Int J Neonatal Screen. 2020 Mar 4;6(1):18. doi: 10.3390/ijns6010018. PMID: 33073015; PMCID: PMC7422974.
10. Farrell, P. M., White, T. B., Ren, C. L., Hempstead, S. E., Accurso, F., Derichs, N., Howenstine, M., McColley, S. A., Rock, M., Rosenfeld, M., Sermet-Gaudelus, I., Southern, K. W., Marshall, B. C., & Sosnay, P. R. (2017). Diagnosis of Cystic Fibrosis: Consensus Guidelines from the Cystic Fibrosis Foundation. The Journal of Pediatrics, 181, S4-S15.e1. https://doi.org/10.1016/j.jpeds.2016.09.064
11. Servidoni MF, Gomez CCS, Marson FAL, Toro AADC, Ribeiro MÂGO, Ribeiro JD, Ribeiro AF; Grupo Colaborativo de Estudos em Fibrose Cística. Sweat test and cystic fibrosis: overview of test performance at public and private centers in the state of São Paulo, Brazil. J Bras Pneumol. 2017 Mar-Apr;43(2):121-128. doi: 10.1590/S1806-37562016000000076. PMID: 28538779; PMCID: PMC5474375.
Related posts:
 DNA fingerprinting: History, Procedure and Its Applications
DNA fingerprinting: History, Procedure and Its Applications
 Epstein-Barr Virus Protein EBNA1 Found to Boost Cancer-Linked Genes in HeLa Cells
Epstein-Barr Virus Protein EBNA1 Found to Boost Cancer-Linked Genes in HeLa Cells
 How Antibiotics Trigger Alpha Satellite DNA Overexpression and Epigenetic Changes
How Antibiotics Trigger Alpha Satellite DNA Overexpression and Epigenetic Changes
 माइकोराइजल फङ्गसले धानमा नाइट्रोजन अवशोषण बढायो
माइकोराइजल फङ्गसले धानमा नाइट्रोजन अवशोषण बढायो
 GABA Breakthrough: Optimizing Anther Culture in Pepper to Accelerate Breeding
GABA Breakthrough: Optimizing Anther Culture in Pepper to Accelerate Breeding

